

Roy M. Long, PhD
Professor; Assistant Dean for Graduate Recruitment
Locations
- Microbiology & Immunology
BSB 252
Contact Information
General Interests
Education
Research Interests

Using genetic, biochemical and cell biological approaches in the yeast S. cerevisiae, my laboratory is interested in studying mechanistic details of RNA localization, a process which post-transcriptionally regulates gene expression. In yeast ASH1 mRNA is localized to the distal end of the daughter cell during anaphase of the cell cycle, resulting in the exclusive deposition of Ash1p in daughter nuclei. Ash1p is a transcriptional repressor, and asymmetric sorting of Ash1p results in asymmetric regulation of transcription.
ASH1 mRNA contains four cis-acting localization elements sufficient to localize a heterologous reporter mRNA to daughter cells. One of these elements resides in the 3’-untranslated region (3’-UTR) while the remaining three elements reside in the coding region of the ASH1 mRNA. Besides these cis-acting elements, five trans-acting factors (SHE1-5) and the actin cytoskeleton are required for localization of ASH1 mRNA. Our laboratory has focused on deciphering mechanistic details related to She1p, She2p and She3p. We demonstrated that the type V myosin, She1p/Myo4p, directly transports ASH1 mRNA containing particles to daughter cells. We also reported that She2p is an RNA-binding protein that directly interacts with each of the ASH1 cis-acting localization elements. Since She3p has the ability to interact with both Myo4p and She2p, we hypothesized that She3p functions to interface Myo4p with the She2p-ASH1 mRNA complex. We demonstrated that a Myo4p-She3p-She2p complex does exist in vivo, and this heterotrimeric complex functions to transport ASH1 mRNA to the bud tip. The function of She3p in ASH1 mRNA localization is not limited to interacting with Myo4p and She2p. We identified mutants of She3p defective for ASH1 mRNA localization, yet these She3p mutants retain the ability to associate with Myo4p and She2p. Our analysis of these She3p mutants suggest that each is defective for associating with ASH1 mRNA, implying that She3p could directly contact the ASH1 cis-acting localization elements. In addition, our experimental evidence suggests that a molecular reorganization of the heterotrimeric complex is required for ASH1 mRNA to be anchored at the bud tip.
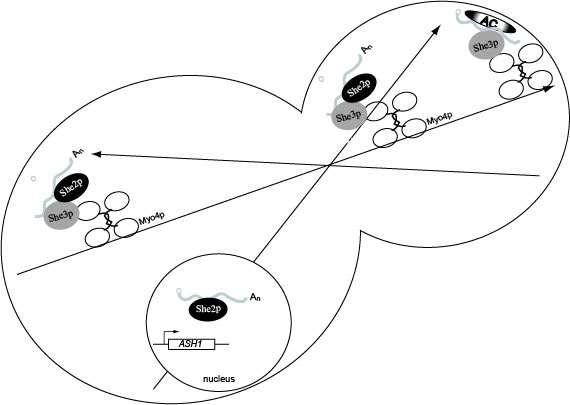
Model for ASH1 mRNA localization. She2p binds ASH1 mRNA co-transcriptionally. In the cytoplasm She3p binds ASH1 mRNA which recruits Myo4p, resulting in formation of the heterotrimeric complex. The heterotrimeric complex is responsible for transporting the cargo to daughter cells on the polarized actin cytoskeleton. Following transport to the bud tip, molecular reorganization of the transport complex results in the release of She2p which allows anchoring factors to associate with ASH1 mRNA, She3p and Myo4p. Once anchored ASH1 mRNA is competent for translation in the daughter cell, and newly synthesized Ash1p is imported into the daughter cell nucleus.
It has been shown that the asymmetric segregation of specific mRNA molecules is imperative for normal development of higher eukaryotic organisms. The mechanisms controlling mRNA transport and localization remain elusive. However, many of the mechanisms controlling other aspects of gene expression such as transcription, translation and splicing, are highly conserved from yeast to man. Consequently, we expect that the mechanisms regulating mRNA localization will also be conserved from yeast to higher eukaryotes. With the power of yeast genetics, we are poised to make significant progress in dissecting the mechanisms controlling mRNA localization.
Supplemental supporting information for:
Gonsalvez GB, Lehmann KA, Ho DK, Stanitsa ES, Williamson JR, Long RM. RNA-protein interactions promote asymmetric sorting of the ASH1 mRNA ribonucleoprotein complex. RNA. 2003 Nov;9(11):1383-99.
Soluble cell lysate preparation
east cells were harvested by centrifugation, and the cell pellet resuspended in lysis buffer: 50 mM HEPES-KOH pH 7.3, 20 mM potassium acetate, 2 mM EDTA, 0.1% Triton X-100, 5% glycerol, 0.1 ug/ml chymostatin, 2 ug /ml aprotinin, 1 ug/ml pepstatin, 0.5 ug/ml leupeptin, and 0.01 ug/ml benzamidine. Glass beads were added to the cell suspension, and cells broken by vortexing. The cell extracts were cleared by centrifugation at 5000 x g for 2 min. at 4oC. The soluble fraction was recovered, mixed with an equal volume of 2X Laemmli buffer and analyzed by Western-blot. Tubulin was detected with alpha-tubulin monoclonal antibody (Serotec) followed by goat anti-rat conjugated with HRP (Roche). Myc fusion proteins were detected as described above.
Generation of polyclonal She2p antiserum
Polyclonal rabbit antiserum was raised against His6-She2p expressed and purified from E. coli. Plasmid pRL266 was transformed into E. coli strain BL21(DE3)pLysS, and expression of His6-She2p was induced with 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG). Following induction, cells were harvested by centrifugation and resuspended in IMAC-5 (20 mM Tris-HCl pH 7.9, 500 mM NaCl, 10% glycerol, 0.1% NP-40 and 5 mM imidazole) containing 175 ug/ml PMSF, 2 ug/ml aprotinin, 2 ug/ml leupeptin, 1 ug/ml pepstatin, 100 ug/ml RNase A and 10 ug/ml DNase I. The cell suspension was passed three times through a French press, and the cell lysate cleared by centrifugation at 30,000 x g for 20 min. at 4oC. His6-She2p was purified from the lysate using Ni-NTA agarose (QIAGEN) and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The portion of the SDS-PAGE gel corresponding to His6-She2p was excised, combined with Freund's adjuvant and injected into a female New Zealand White rabbit.
Expression and purification of recombinant GST-She2p and untagged She2p
5mlLB+ampicillin+chloramphenicol cultures of E. coli BL21(DE3)pLysS transformed with pGEX-2T, pRL172, pRL270 or pRL273 were grown overnight at 37oC. The overnight cultures were diluted into 100 ml of fresh media resulting in an initial OD600 of 0.05. The cultures were grown to an OD600 of 0.5 at 30oC, induced with 1 mM IPTG and incubated at 30oC for 3 hr. with vigorous shaking. Cells were harvested by centrifugation and the cell pellet resuspended in 1X phosphate buffered saline containing 1% Triton X-100, 0.1 ug/ml chymostatin, 2 ug/ml aprotinin, 1 ug/ml pepstatin, 0.5 ug/ml leupeptin, and 0.01 ug/ml benzamidine. Cell lysates were prepared by passing the cell suspension four times through a French press, and the soluble fraction recovered after centrifugation at 15,000 x g for 15 min. at 4oC. The GST fusion proteins were purified using glutathione sepharose (Pharmacia) according to the manufacturer's directions.
For in vitro experiments using untagged She2p, the ORF corresponding to She2p was cloned into the BamHI site of plasmid pHMTc. This plasmid is a derivative of pMal-c2x (New England Biolabs) which replaces the factor Xa protease recognition site with a TEV cleavage site and adds a His6-epitope tag directly upstream of the maltose binding (MBP) coding sequence. Expression of MBP-She2p was induced with 1 mM IPTG. Two grams of E. coli BL21(DE3) cells expressing MBP-She2p were disrupted by sonication in 100 ml of a buffer containing 20 mM Tris-HCl pH 7.5, 200 mM NaCl, 10 mM EDTA, 1 mM DTT and two complete EDTA-free protease inhibitor tablets (Roche). After clarification of the lysate by centrifugation (27,000 x g for 20 min), MBP-She2p was purified by affinity chromatography on an amylose column according to the manufacturer's recommendations (New England Biolabs). Selected fractions from the amylose column were pooled and concentrated five-fold using a YM30 Centriprep Centricon (Millipore), and the MBP-She2p fusion protein was cleaved using TEV protease (Gibco-BRL) to liberate free She2p. The proteolyzed sample was dialyzed against 50 mM Tris-HCl pH 7.5, 10 mM KCl and 1mM DTT, and free She2p was purified from MBP by POROS HQ anion exchange chromatography on a BioCAD SPRINT (Perceptive Biosystems). The HQ column was developed in a KCl gradient in a buffer containing 50 mM Tris-HCl pH 7.5 and 1 mM DTT. Eluted fractions containing She2p were identified by SDS-polyacrylamide gel electrophoresis. The final She2p pool was concentrated on a BioCAD SPRINT HQ column prior to dialysis against 20 mM potassium phosphate pH 7.4, 20 mM NaCl and 1 mM DTT, and stored at 4 oC. The yield of She2p was approximately 10 mg per liter of cells, and the preparation of She2p was determined to be greater than 98% pure by coomassie blue stained SDS-PAGE and MALDI-TOF mass spectrometry.
In vitro transcription
32P-labeled RNAs for UV cross-linking assays were prepared by in vitro transcription. For RNA corresponding to the ASH1 E3 cis-acting localization element plasmid pRL168 was linearized with either HindIII or PstI and subsequently used for in vitro transcription. For MS2 RNA plasmid pRL679 was linearized with HindIII prior to in vitro transcription. For UV cross-linking experiments, in vitro transcription reactions were performed with T7 or SP6 RNA polymerases and alpha-32P-UTP (Perkin Elmer). Labeled RNAs were purified by polyacrylamide gel electrophoresis on 6% denaturing gels.
For electrophoretic mobility shift assays, RNA was prepared by run-off in vitro transcription with T7 RNA polymerase. RNAs were purified on a 10% denaturing gel, visualized by UV shadowing, excised and eluted into 20 mM Tris pH 6.8, 1 M NaCl and 1 mM EDTA. Purified RNAs were precipitated in ethanol and stored in deionized water at —20 oC. A portion (5 pmol) of the gel purified RNA was treated with calf intestinal alkaline phosphatase and labeled at its 5' end using 6000 Ci/mmol [gamma 32P] ATP and T4 polynucleotide kinase, and purified from a second denaturing gel prior to use.
In situ hybridization and immunofluorescence
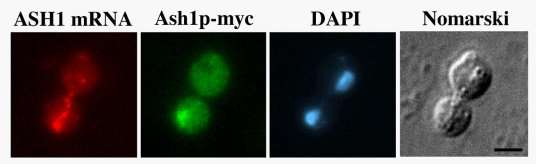
In situ hybridization and immunofluorescence were performed as described previously (Long et al., 1997). Briefly, yeast cells were grown in appropriate media to mid-log phase, fixed with formaldehyde and converted to spheroplasts. For in situ hybridization Cy3 conjugated ASH1 or lacZ DNA oligonucleotides probes were used (Long et al., 1997). For immunofluorescence anti-myc monoclonal antibody 9E10 (Roche) was used followed by FITC-conjugated goat-anti mouse IgG (Jackson ImmunoResearch Laboratories, Inc.). Cells were subsequently stained with 4',6'-diamidino-2phenylindole (DAPI) and mounted on slides with mounting media containing phenylenediamine.
Images were captured using a Nikon Eclipse 600 epifluorescence microscope equipped with a 60X N.A. 1.4 objective, interfaced to a Micromax-Interline Transfer CCD Camera (Princeton Instruments, Inc.) and MetaMorph Imaging Software (Universal Imaging Corp.).
Site directed mutagenesis
The QuikChangeTM Site-Directed Mutagenesis Kit (Stratagene) was used according to manufacturer's direction to generate She2p mutants R43K, R44K, R49K and R52K. Alternatively, She2p mutants R43A, R44A, R49A, R52A and R63A were generated by overlap PCR (Clackson et. al. 1991). Briefly, four primers were used per mutant in two sequential rounds of PCR. In the first round of PCR, two overlapping PCR fragments were generated containing the mutated sequence within the region of overlap between the two PCR products. These two PCR fragments were purified and served as templates for a second PCR reaction to amplify SHE2. All mutations in the SHE2 open reading frame were confirmed by DNA sequencing and She2p expression was confirmed by Western-blotting. Information for the oligonucleotides used for overlap PCR are available upon request.
Publications
-
(Gonyo P, Bergom C, Brandt AC, Tsaih SW, Sun Y, Bigley TM, Lorimer EL, Terhune SS, Rui H, Flister MJ, Long RM, Williams CL.) Oncogene. 2017 Dec 14;36(50):6873-6883 PMID: 28806394 PMCID: PMC5730474 SCOPUS ID: 2-s2.0-85038242723 08/15/2017
-
Spatial regulation of translation through RNA localization.
(Gonsalvez GB, Long RM.) F1000 Biol Rep. 2012;4:16 PMID: 22912650 PMCID: PMC3412389 08/23/2012
-
Techniques for following the movement of single RNAs in living cells.
(Urbinati CR, Long RM.) Wiley Interdiscip Rev RNA. 2011;2(4):601-9 PMID: 21957047 SCOPUS ID: 2-s2.0-84855291135 10/01/2011
-
She3p possesses a novel activity required for ASH1 mRNA localization in Saccharomyces cerevisiae.
(Landers SM, Gallas MR, Little J, Long RM.) Eukaryot Cell. 2009 Jul;8(7):1072-83 PMID: 19429778 PMCID: PMC2708451 SCOPUS ID: 2-s2.0-67650457062 05/12/2009
-
Nonsense-mediated decay of ash1 nonsense transcripts in Saccharomyces cerevisiae.
(Zheng W, Finkel JS, Landers SM, Long RM, Culbertson MR.) Genetics. 2008 Nov;180(3):1391-405 PMID: 18791219 PMCID: PMC2581943 SCOPUS ID: 2-s2.0-59449110397 09/16/2008
-
Monitoring the temporal and spatial distribution of RNA in living yeast cells.
(Long RM, Urbinati CR.) Methods Mol Biol. 2008;419:187-96 PMID: 18369984 SCOPUS ID: 2-s2.0-42949178004 03/29/2008
-
Loc1p is required for efficient assembly and nuclear export of the 60S ribosomal subunit.
(Urbinati CR, Gonsalvez GB, Aris JP, Long RM.) Mol Genet Genomics. 2006 Oct;276(4):369-77 PMID: 16871394 SCOPUS ID: 2-s2.0-33748319405 07/28/2006
-
(Gallas MR, Dienhart MK, Stuart RA, Long RM.) Mol Biol Cell. 2006 Sep;17(9):4051-62 PMID: 16790493 PMCID: PMC1556384 SCOPUS ID: 2-s2.0-33748108494 06/23/2006
-
RNA localization in yeast: moving towards a mechanism.
(Gonsalvez GB, Urbinati CR, Long RM.) Biol Cell. 2005 Jan;97(1):75-86 PMID: 15601259 SCOPUS ID: 2-s2.0-13544261683 12/17/2004
-
ASH1 mRNA anchoring requires reorganization of the Myo4p-She3p-She2p transport complex.
(Gonsalvez GB, Little JL, Long RM.) J Biol Chem. 2004 Oct 29;279(44):46286-94 PMID: 15328357 SCOPUS ID: 2-s2.0-8544254751 08/26/2004
-
RNA-protein interactions promote asymmetric sorting of the ASH1 mRNA ribonucleoprotein complex.
(Gonsalvez GB, Lehmann KA, Ho DK, Stanitsa ES, Williamson JR, Long RM.) RNA. 2003 Nov;9(11):1383-99 PMID: 14561888 PMCID: PMC1287060 SCOPUS ID: 2-s2.0-0142240402 10/17/2003
-
The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU.
(Sato H, Frank DW, Hillard CJ, Feix JB, Pankhaniya RR, Moriyama K, Finck-Barbançon V, Buchaklian A, Lei M, Long RM, Wiener-Kronish J, Sawa T.) EMBO J. 2003 Jun 16;22(12):2959-69 PMID: 12805211 PMCID: PMC162142 SCOPUS ID: 2-s2.0-0038376087 06/14/2003