

Martin J. Hessner, PhD
Professor, Pediatrics (Endocrinology); Director, Max McGee Research Center for Juvenile Diabetes
Locations
- Pediatrics (Endocrinology)
TBRC C2530
Contact Information
General Interests
Education
BS, Carroll College, Waukesha, WI, 1985
Research Interests
Type 1 diabetes (T1D) is an autoimmune disease that targets the pancreatic beta-cell. It is among the most common chronic childhood illness. T1D is a complex disease, meaning that it involves both genetic determinants as well as environment factors. In recent decades T1D is being diagnosed at an earlier age with an increasing incidence in most developed countries. This points towards increasing environmental pressure. It is known that T1D pathogenesis involves the expansion of autoreactive T-cells and B-cells. It also involves alterations in innate immune activity that are poorly understood.
The study of T1D has many challenges. Relevant tissues, such as pancreas and pancreatic lymph nodes are inaccessible from living patients. Cells and immune mediators associated with T1D are dilute in the periphery and difficult to detect. Further, a focus on one or a few mediators may be uninformative due to important combinatorial effects. To circumvent these obstacles, we have optimized a bioassay where patient plasma is co-cultured with a well-controlled ‘reporter’ cell population. The induced transcriptional signature is then globally measured with a microarray or through RNAseq, subjected to ontological analyses, and used to direct follow-up studies. This noninvasive approach enables an integrated functional assessment of the many plasma components (cytokines, chemokines, metabolites, microbial antigens). It allows for the sensitive detection and differentiation of immune states. This has fostered new insight in our studies of T1D.
Our laboratory uses the BioBreeding (BB) rat model of T1D. In the BB rat we have identified an underlying, partially interleukin-1 (IL-1) dependent, inflammatory state that is associated with T1D susceptibility, yet is independent of T1D progression. This state includes elevated plasma chemokine levels and beta-cell phenotypes consistent with microbial antigen exposure and pattern recognition receptor activation. BB DR (diabetes resistant) rats do not develop spontaneous diabetes; however, T1D can be triggered in young rats T1D by viral infection. Notably, we have discovered that the innate state in DR rats is temporally supplanted by an age-dependent IL-10/TGF-β-mediated regulatory state that coincides with the inability of virus to trigger T1D in older rats.
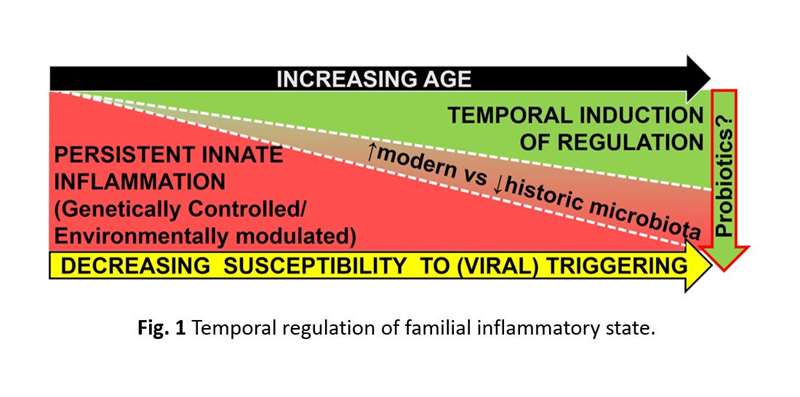
Similarly, our studies of diabetes patients, their healthy siblings and unrelated controls have also identified an innate inflammatory state in T1D families. This state is also consistent with pattern recognition receptor activation and includes elevated plasma cytokine levels, yet is independent of the high-risk HLA genotypes, autoantibody status, and T1D progression. Notably, among sibling non-progressors, the innate state is temporally supplanted by an IL-10/TGF-β-mediated regulatory state that includes increases in peripheral activated regulatory T-cell (Treg) frequencies. Our studies suggest that subclinical T1D progression is promoted when the underlying innate state is augmented by additional inflammatory signals (e.g. viral infection) prior to the establishment of protective regulatory mechanisms (Figure 1). This provides possible insight as to why T1D often develops in children and adolescents.
Our latest studies find that a higher innate inflammatory bias at T1D onset is predictive of 1) an accelerated rate of decline in post-onset β-cell function in newly diagnosed patients, and 2) greater responsiveness to immunotherapy at clinical onset. Since retention of even modest levels of β-cell function reduces the risk for microvascular complications and hypoglycemic events, gaining an understanding the variables that govern T1D progression is significant.
The rise in T1D incidence coincides with environmental changes that have likely altered the gut microbiota. These changes include increased antibiotic usage and introduction of the Western diet. Further, dysbiosis and intestinal hyper-permeability have been associated with T1D in rodent models and humans. Notably, we find that prebiotic and probiotic approaches that alter the composition of the GI microbiota can partially normalize the innate inflammatory state in BB rats and prevent T1D progression. We have also determined that probiotic supplement can partially normalize the familial inflammatory state in siblings of T1D patients.
Based on these observations, the Hessner Laboratory has two major ongoing initiatives:
The first area of focus is characterization of the immunological basis of the familial inflammation associated with T1D susceptibility, how levels of innate inflammation impact the rate of disease progression, and how variation in inflammation among new onset patients impacts the response to immunotherapy.
The second area of focus is how environmental factors, such as probiotics, may be leveraged to promote immunoregulation and possibly slow T1D progression before and at T1D onset.
Our long-term goal is to foster the development of disease modifying strategies in those with or at high risk of developing T1D, either using probiotics alone or in combination with other therapeutic agents.
Relevant Publications:
Innate immune activity as a predictor of persistent insulin secretion and association with responsiveness to CTLA4-Ig treatment in recent-onset type 1 diabetes. (Cabrera SM, Engle S, Kaldunski M, Jia S, Geoffrey R, Simpson P, Szabo A, Speake C, Greenbaum CJ, Type 1 Diabetes TrialNet CTLA4-Ig (Abatacept) Study Group, Chen YG, Hessner MJ.) Diabetologia. 2018 11;61(11):2356-2370.
Modulation of the diet and gastrointestinal microbiota normalizes systemic inflammation and β-cell chemokine expression associated with autoimmune diabetes susceptibility. (Henschel AM, Cabrera SM, Kaldunski ML, Jia S, Geoffrey R, Roethle MF, Lam V, Chen YG, Wang X, Salzman NH, Hessner MJ.) PLoS One. 2018;13(1):e0190351.
Blood-based signatures in type 1 diabetes. (Cabrera SM, Chen YG, Hagopian WA, Hessner MJ.) Diabetologia. 2016 Mar;59(3):414-25.
Interleukin-1 antagonism moderates the inflammatory state associated with Type 1 diabetes during clinical trials conducted at disease onset. (Cabrera SM, Wang X, Chen YG, Jia S, Kaldunski ML, Greenbaum CJ, Type 1 Diabetes TrialNet Canakinumab Study Group, Mandrup-Poulsen T, AIDA Study Group, Hessner MJ.) Eur J Immunol. 2016 Apr;46(4):1030-46.
Innate inflammation in type 1 diabetes. (Cabrera SM, Henschel AM, Hessner MJ.) Transl Res. 2016 Jan;167(1):214-27. Molecular signatures differentiate immune states in type 1 diabetic families. (Chen YG, Cabrera SM, Jia S, Kaldunski ML, Kramer J, Cheong S, Geoffrey R, Roethle MF, Woodliff JE, Greenbaum CJ, Wang X, Hessner MJ.) Diabetes. 2014 Nov;63(11):3960-73.
Publications
-
Impaired taurine transport contributes to bronchopulmonary dysplasia: a systems biology analysis.
(Jing X, Wang Y, Roethle M, Jia S, Teng M, Konduri GG, Hessner MJ, Pritchard KA Jr, Day BW, Jin Y, Ma X, Naylor S, Lin CW, Gu H, Du J, Teng RJ.) Am J Respir Cell Mol Biol. 2026 Aug 01;74(8):1058-1072 PMID: 41738245 02/25/2026
-
(Bedrat A, Truchan NA, Pant T, Jia S, Roethle MF, Chen YG, Lin CW, Hessner MJ.) Diabetologia. 2026 Jul 11 PMID: 42432282 SCOPUS ID: 2-s2.0-105044473999 07/11/2026
-
(Ciecko AE, Nabi R, Drewek A, Schauder DM, Zakharov PN, Wan X, Lieberman SM, Cui W, Hessner MJ, Lin CW, Chen YG.) iScience. 2025 Oct 17;28(10):113537 PMID: 41050931 PMCID: PMC12494592 10/06/2025
-
(Mucalo Katunaric L, Jia S, Singh A, Roethle MF, Panepinto JA, Brousseau DC, Hessner MJ, Brandow AM.) Blood Adv. 2025 Aug 12;9(15):3790-3800 PMID: 40238896 PMCID: PMC12309595 SCOPUS ID: 2-s2.0-105012219602 04/16/2025
-
(Kozlovich SY, Pan AY, McIntosh JJ, Palatnik A, Jia S, Hessner MJ, Nghiem-Rao TH.) Sci Rep. 2025 Jul 25;15(1):27041 PMID: 40715324 PMCID: PMC12297459 SCOPUS ID: 2-s2.0-105011750510 07/28/2025
-
(Ciecko AE, Nabi R, Drewek A, Schauder DM, Zakharov PN, Wan X, Lieberman SM, Cui W, Hessner MJ, Lin CW, Chen YG.) Iscience. 17 October 2025;28(10) SCOPUS ID: 2-s2.0-105016644538 10/17/2025
-
(Pant T, Lin CW, Bedrat A, Jia S, Roethle MF, Truchan NA, Ciecko AE, Chen YG, Hessner MJ.) Sci Adv. 2024 May 17;10(20):eadn2136 PMID: 38758799 PMCID: PMC11100571 SCOPUS ID: 2-s2.0-85193631768 05/17/2024
-
Cellular Senescence Contributes to the Progression of Hyperoxic Bronchopulmonary Dysplasia.
(Jing X, Jia S, Teng M, Day BW, Afolayan AJ, Jarzembowski JA, Lin CW, Hessner MJ, Pritchard KA Jr, Naylor S, Konduri GG, Teng RJ.) Am J Respir Cell Mol Biol. 2024 Feb;70(2):94-109 PMID: 37874230 PMCID: PMC12042139 SCOPUS ID: 2-s2.0-85184825553 10/24/2023
-
(Zhang X, Moore CM, Harmacek LD, Domenico J, Rangaraj VR, Ideozu JE, Knapp JR, Woods KJ, Jump S, Jia S, Prokop JW, Bowler R, Hessner MJ, Gelfand EW, Levy H.) JCI Insight. 2022 Mar 22;7(6) PMID: 35315363 PMCID: PMC8986072 SCOPUS ID: 2-s2.0-85126883712 03/23/2022
-
Characterizing T cell responses to enzymatically modified beta cell neo-epitopes.
(Nguyen H, Arribas-Layton D, Chow IT, Speake C, Kwok WW, Hessner MJ, Greenbaum CJ, James EA.) Front Immunol. 2022;13:1015855 PMID: 36703975 PMCID: PMC9871889 SCOPUS ID: 2-s2.0-85146920361 01/28/2023
-
(Sargin P, Roethle MF, Jia S, Pant T, Ciecko AE, Atkinson SN, Salzman NH, Teng RJ, Chen YG, Cabrera SM, Hessner MJ.) Gut Microbes. 2022;14(1):2136467 PMID: 36261888 PMCID: PMC9586621 SCOPUS ID: 2-s2.0-85140364986 10/21/2022
-
(Pritchard KA Jr, Jing X, Teng M, Wells C, Jia S, Afolayan AJ, Jarzembowski J, Day BW, Naylor S, Hessner MJ, Konduri GG, Teng RJ.) PLoS One. 2022;17(8):e0269564 PMID: 36018859 PMCID: PMC9417039 SCOPUS ID: 2-s2.0-85137126858 08/27/2022