
Study Reveals a Link Between Thrombophilia, Placental Abruption, and Obstetric Hemorrhage
Milwaukee, Nov. 6, 2020 -- The placenta plays key roles in supporting fetal growth and regulating maternal physiology. Abnormal placentation can result in miscarriages and stillbirth, and obstetric syndromes of preterm birth and preeclampsia. In human placentation, fetal villi containing fetal blood vessels are bathed in maternal blood. The outermost layers of these villi are made up of specialized cells called trophoblast cells. These cells also invade deep into maternal tissues. They invade spiral arteries and to a lesser extent veins, replacing maternal endothelial cells. This anatomic location is predicted to give trophoblast cells a unique ability to regulate local vascular functions in uteroplacental circulation. This type of placentation where fetal cells have direct access to maternal blood is called hemochorial placentation and is also found in rats and mice.
Thrombophilia is an inherited or acquired abnormality that increases an individual’s tendency to form blood clots. The unusual extravascular blood flow of hemochorial placentation is thought to further increase this tendency and cause placenta-mediated pregnancy complications in women with preexisting thrombophilia. Yet, causation between thrombophilia and placental insufficiency has remained difficult to prove. Hallmarks of pathological thrombosis in uteroplacental circulation have also remained poorly defined. In a study published in Science Advances today, researchers at the Medical College of Wisconsin used murine models of clotting factor and platelet function defects to identify factors that might precipitate placental thrombosis and cause adverse pregnancy outcomes due to maternal or fetal thrombophilia.
The study utilized mice that lack an anticoagulant protein, the Endothelial Protein C Receptor (EPCR). Embryos with homozygous deficiency of this protein exhibit placental insufficiency and do not survive past midgestation. Using a variety of genetic tools, the researchers found that preventing thrombin-mediated activation of maternal platelets allowed placental development and in utero survival of mice lacking EPCR. However, about half of the pups did not survive past the neonatal period. An examination of the placenta revealed persistent blood clots in maternal venous channels of uteroplacental circulation and a remarkable absence of pathology in fetal placenta. The researchers noted that neonatal death and placental venous thrombosis were not observed when EPCR expression was maintained on trophoblast cells but removed in the embryo.
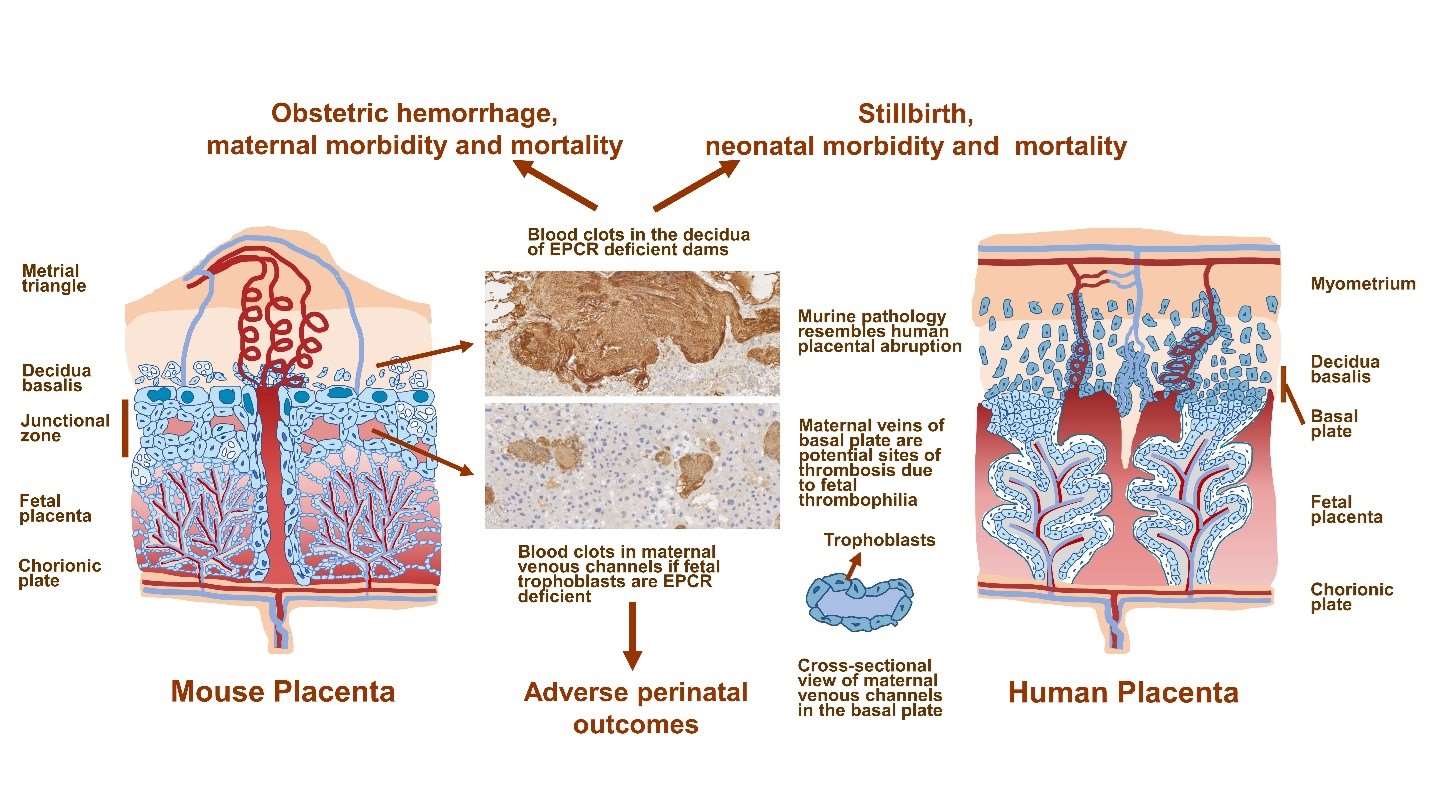
In the course of these studies, the researchers made another unexpected discovery. They observed that female mice that lacked the gene for EPCR exhibited high incidences of pregnancy-induced vaginal bleeding, pregnancy loss, stillbirth, and maternal death. Surgical and pathological evaluation of pregnancies revealed extensive hemorrhaging in the uterus, specially in regions where the placenta is attached to maternal tissues. Large blood clots were readily observed in these regions. These blood clots were characterized by immune cell infiltration and features of NETosis, a form of immune cell death. The researchers finally showed that if EPCR-deficient dams lack Par4, a widely expressed thrombin receptor, pathological blood clotting and its sequel is averted. They note that further experimentation will be necessary to determine whether pharmacological targeting of thrombin receptors can effectively prevent or reverse pathology. The authors plan to use this murine model of maternal thrombophilia-associated placental abruption to identify cellular and molecular interactions that lead to immunothrombosis and to look for associated biomarkers.
What does this study mean for maternal and fetal health?
The authors interpret these as proof-of-concept studies that have confirmed the long-suspected link between maternal thrombophilia and placental abruption, a life- threatening condition for the mother and the fetus. Placental abruption refers to premature separation of the placenta from maternal tissues due to local hemorrhaging. It is unclear whether this effect is specific to EPCR deficiency or other thrombophilia mutations might have a similar effect. Authors note that EPCR has been reported to be shed from membrane surfaces in inflammatory and infectious conditions and in association with fetal death.
The murine study shows that placental abruption is characterized by pathological changes that occur long before its acute presentation. This provides hope that it may be possible to identify predictive biomarkers and therapies to prevent or reverse disease. The study indicates that targeting thrombin receptor Par4 might provide benefit in some scenarios. Much more research is needed to test these possibilities. This is one more exciting step towards unraveling the complex biology of pregnancy and placentation.
Keep up with the latest news. Sign up for Newsroom Alerts.
Latest press releases, stories and resources.
MCW Media Contacts
The media relations team at MCW is happy to assist in coordinating experts for interviews. Please reach out to us at:
media@mcw.edu
(414) 955-8764
Contact us
