
Andreas Beyer Lab

Andreas M. Beyer, PhD

Curriculum Vitae (PDF)
X: @BeyerLab
abeyer@mcw.edu
Andreas M. Beyer is a professor of medicine and physiology at the Medical College of Wisconsin and the co-director of the basic and translational research program in cardio-oncology.
During his training in genetics and physiology, he has gained detailed expertise in generating and evaluating novel approaches in genetics, molecular biology and physiology. In his time spent in the lab he performs experimental troubleshooting involving video microscopy, fluorescent microvascular imaging, generation of genetic rodent models, physiological evaluation of in vivo vascular function and blood pressure. With the support of this research group and important local and national collaborators, the Beyer lab is using live human tissues to address important questions in vascular biology that will lead to clinically relevant findings and drive further exploration of mechanism in rodent models. His lab hopes that clinically relevant data from human tissues will enable a detailed mechanistic understanding of disease that can then be used to develop novel therapeutics and translate both diagnostics and therapies themselves to the clinic.
Lab Projects
Our collaborative team has established that the mechanisms of flow-mediated dilation (FMD) changes over the lifespan and shift with the onset of CAD. In healthy patients, FMD is regulated by the vasoprotective dilator nitric oxide (NO). In contrast, in CAD patients or in vessels exposed to acute stressors, NO bioavailability is reduced, and FMD is attributed to a compensatory rise in hydrogen peroxide (H2O2), a pro-inflammatory reactive oxygen species (ROS). These findings expanded our understanding of CAD, which was previously viewed as a disease limited to large conduit arteries, to reflect microvascular pathology to with significant prognostic implications. Our studies pioneered genetic manipulation techniques (siRNA, viral overexpression) in the human coronary circulation to answer mechanistic questions.
Work in my lab has long integrated animal studies with mechanistic evaluation of isolated human microvessels. Recent collaborative projects led us to expand our repertoire to study vascular function in vivo (e.g., brachial artery FMD, arterial tonometry, echocardiography, skin microdialysis/laser Doppler flowmetry). Our work fulfills a critical need to translate preclinical findings into human studies and thus serves as a bridge to clinical investigations. Understanding the molecular and physiological changes that contribute to CVD has clinical implications that, may lead to novel means to predict pathological changes and intervene before irreversible damage occurs.
Current work in understanding the pathological changes that occur with onset of CAD focuses on the role of TERT, the catalytic subunit of telomerase, in maintaining vasodilator function in the human microcirculation. Specifically, we study a noncanonical role of TERT in preserving mitochondrial homeostasis. We discovered previously unrecognized signaling between TERT and well-known regulators of vascular health, such as the renin angiotensin system and autophagy. Data from my lab and that of others implicate decreased levels of TERT with elevated mitochondrial DNA (mtDNA) damage as important regulators of mitochondrial integrity. This evidence led to a new collaborative work designed to understand the contribution of mitochondrial networks and their regulation to pathological changes in the human microcirculation. In specific, we aim to define how mitochondrial fission and fusion and its regulation, a fundamental process to maintain mitochondrial and cellular health, is critically linked as a regulator of FMD in the human microcirculation.
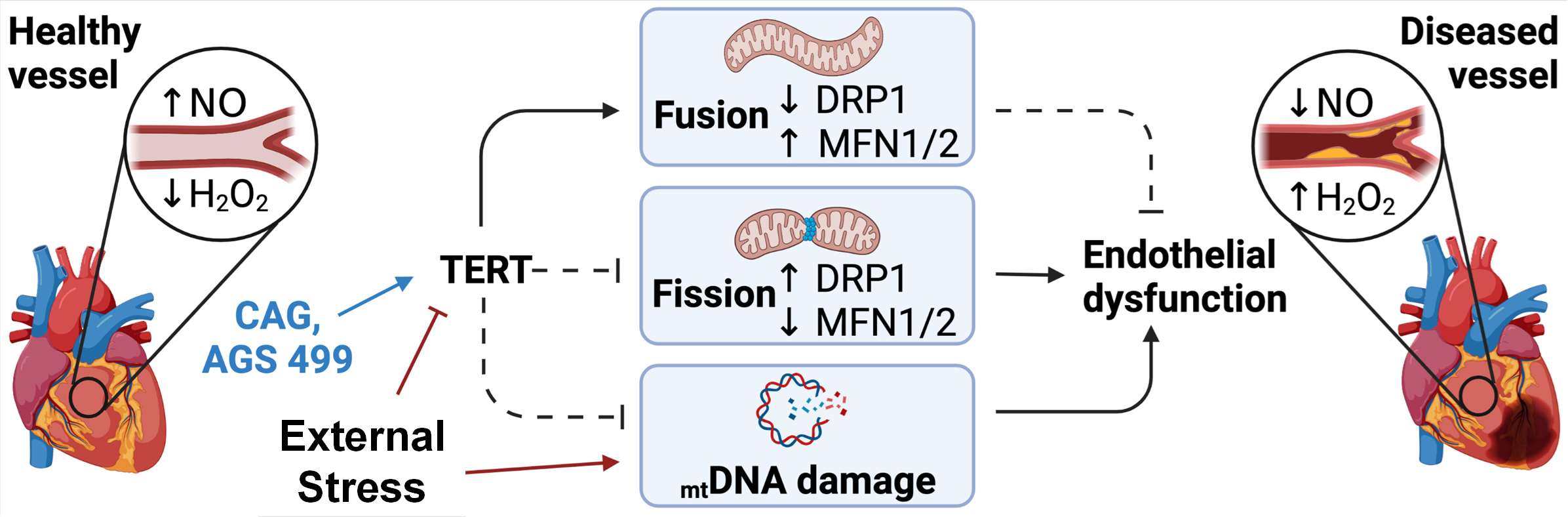
Fig. 1. Mitochondrial integrity and microvascular function.
Cardiovascular disease (CVD) and cancer are the number one and two causes of mortality and morbidity, and it is increasingly recognized that cancer and CVD share overlapping risk profiles. Long-term cancer survival is closely tied to CVD, while CVD conditions contribute to the progression of cancer and influence relevant treatment choices. With the use of systemic and targeted anti-cancer therapies (CTx), the number of annual deaths from cancer has been significantly reduced; however, most CTx agents have severe adverse consequences for the cardiovascular system. In fact, CVD related to CTx has emerged as the leading cause of non-cancer related deaths among breast cancer (BC) survivors. Given the magnitude of the clinical problem of cardiovascular-related mortality in cancer survivors, the novel clinical field of cardio-oncology has emerged with the aim of improving long-term, disease-free survival in cancer patients while addressing the underlying mechanism of cardiovascular comorbidity. While injury to the heart resulting from exposure to CTx is well established, little data exist on the contribution of CTx to endothelial cell dysfunction and the direct effect on mitochondria in the microcirculation. Microvascular endothelial dysfunction is the best predictor of future cardiovascular events, superior even to the degree of large vessel disease (i.e., CAD (coronary artery disease)) or ejection fraction in patients with or without coronary stenoses. This suggests that understanding the long-term consequences of CTx on microvascular function represents a novel, to date mostly unexplored, avenue to understanding and predicting the risk of cardiovascular complications in cancer patients.
We are using lessons learned from our ongoing investigations in understanding the changes in microvascular function in patients with CAD and expand them to investigate the vascular and mitochondrial changes and systemic consequences in cancer patients undergoing clinical necessary anti-cancer therapy. The objectives of this proposal are to 1) establish the contribution of microvascular dysfunction to CVD progression with an emphasis on cardio-oncology; 2) Establish the relevance of mitochondrial damage and secondary signaling in development and progression of CV events in cancer survivors; 3) define and predict risk for adverse CV events in cancer patients building on concepts of systems biology.
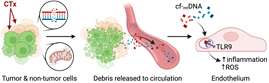
Fig. 3. CTx-induced mitochondrial damage and secondary signaling.
Current Members

Laura Norwood Toro
Research Scientist II
lnorwood@mcw.edu
Laura Norwood Toro is a research scientist in the Andreas Beyer Lab. Her primary responsibility is to explore the effect of chemotherapy on cardiovascular outcomes in coronary circulation and vascular endothelium. One focus of her studies is to investigate the functions of telomerase in the nucleus versus the mitochondria. Her background is in cell biology and molecular biology.

Steve Hammond, PhD
Postdoctoral Researcher
Steve Hammond is a postdoctoral researcher in the Beyer lab. Prior to his start at MCW, he earned his doctorate from Kansas State University where he investigated the adverse cardiovascular consequences of 5-fluorouracil chemotherapy and modalities to alleviate their occurrence. Steve is most interested in better understanding the mitochondrial contributions to cardiovascular function in health in disease. He is currently studying the role of mitochondrial signaling in the development of anticancer therapy induced microvascular dysfunction. Specifically, his primary projects aim to elucidate the role of tumor derived circulating factors and alterations in mitochondrial calcium signaling in the development of microvascular pathology during anti-cancer treatment.

Erin Birch
Research Technologist II
ebirch@mcw.edu
Erin Birch is a research technologist in Dr. Beyer and Dr. Zhang’s labs. Erin has experience researching autoimmune diseases, chronic kidney disease, and cardiovascular disease. She is working with Dr. Beyer’s lab to provide support with several projects. Erin also contributes to the analysis of microvascular function in patients with coronary artery disease, COVID-19, and healthy patients. In her free time, Erin enjoys traveling and adventuring in the Rocky Mountains.
Lukas Brandt
Research Assistant
lbrandt@mcw.edu
Lukas Brandt is a graduate student in Dr. Beyer's lab. Prior to starting at MCW's physiology program, he received his bachelor's degree from the University of Applied Science Bingen, Germany with a major in biotechnology. He received additional training in a Molecular Biology master program at the Goethe University Frankfurt, Germany before pursuing his education in physiology at MCW. His research interests are whole organ physiology with a focus on cardiovascular pathology in response to clinical used anti-cancer therapy. Lukas aims to utilize rodent models to investigate physiological and molecular changes that lead to cardiovascular disease.

Cristhian Gutierrez Huerta
Research Assistant Graduate School
cgutierrez@mcw.edu
Cristhian Gutierrez Huerta is a graduate MSTP student in the Beyer/Gutterman Lab. He graduated with a BS in Applied Mathematics and Biological Sciences from the University of California, Merced in 2018. He then completed a 2-yr post-baccalaureate research fellowship at the National Heart, Lung, and Blood Institute. In the Beyer/Gutterman group, he will begin work on identifying the role of mitochondrial fission/fusion on microvascular endothelial function and its relationship to coronary artery disease progression.

Yoshinori Nishijima
Research Associate
ynishijima@mcw.edu
My current research is focused on understanding the transient receptor potential vanilloid 4 channels to angiotensin II-induced impairment of vasodilation in animals and humans. I am also interested in the role of vascular smooth muscle potassium channels in hydrogen peroxide-induced vasodilation in human arterioles, in health and disease.
Alumni/Former Trainees
- Karima Ait-Aissa
- Karen Clark, PhD
- Daniela Didier
- Johnathan Ebben
- Alena Hanson
- Joe Hockenberry
- Bill Hughes, PhD
- Andrew D. Kadlec, PhD
- Minhi Kang
- Todd Le
- Jasmine Linn
- Janée Terwoord
- Micaela Young
Recent Publications
-
(Hammond ST, Brandt L, Kong AL, White SB, Lewandowski D, Beyer AM.) Arterioscler Thromb Vasc Biol. 2026 Jun;46(6):e319865 PMID: 41924873 PMCID: PMC13048292 SCOPUS ID: 2-s2.0-105035117044 04/02/2026
-
Guidelines for evaluating endothelial function in vascular tissue.
(McCarthy CG, Aalkjær C, Bagher P, Beyer AM, Boedtkjer E, Bomfim GF, Breslin JW, Briones AM, Castorena-Gonzalez JA, Costa TJ, Dai Z, Davel AP, Earley S, Freed JK, Garland C, Isakson BE, Jepps TA, Kalucka J, Lavanderos B, Makino A, Norton CE, Segal SS, Tan W, Trask AJ, Wilson C, Zawieja SD, Wenceslau CF.) Am J Physiol Heart Circ Physiol. 2026 May 01;330(5):H1600-H1672 PMID: 41801061 PMCID: PMC13085230 03/09/2026
-
Telomer length, integrity vs. telomerase-activity: who is to blame for heart failure?
(Kleinbongard P, Beyer AM.) Cardiovasc Res. 2026 Mar 26;122(4):433-435 PMID: 41634935 PMCID: PMC13020426 SCOPUS ID: 2-s2.0-105034456796 02/04/2026
-
Molecular Lung Imaging Following Exposure to Radiation Predicts Long-Term Survival in Rats.
(Clough AV, Mpala K, Taheri P, Toro LN, Beyer AM, Gasperetti T, Zhao M, Kerns S, Himburg HA, Audi SH.) Int J Mol Sci. 2026 Mar 08;27(5) PMID: 41828702 PMCID: PMC12986413 SCOPUS ID: 2-s2.0-105032664518 03/14/2026
-
A comprehensive multi-organ proteomic atlas of human aging across 50 years
(Dong W, Klosa P, Beyer A, Lin X, Liu C.) Protein and Cell. April 2026;17(4):275-278 SCOPUS ID: 2-s2.0-105037024468 04/01/2026
-
Autophagy modulates the mechanism of flow-mediated dilation upstream of telomerase.
(Hughes WE, Hader SN, Astbury K, Brandt L, Ait-Aissa K, Gutterman DD, Beyer AM.) Basic Res Cardiol. 2025 Dec;120(6):1131-1140 PMID: 41238787 PMCID: PMC12680695 SCOPUS ID: 2-s2.0-105021839661 11/15/2025
-
(Venkatasubramanian R, Mahoney SA, Hutton DA, VanDongen NS, Brunt VE, Greenberg NT, Longtine AG, Brandt L, Beyer AM, Melov S, Rossman MJ, Seals DR, Clayton ZS.) Am J Physiol Heart Circ Physiol. 2025 Dec 01;329(6):H1672-H1683 PMID: 41143747 PMCID: PMC12704655 SCOPUS ID: 2-s2.0-105024255414 10/27/2025
-
(Terwoord JD, Norwood Toro LE, Hader SN, Hammond ST, Hockenberry JC, Linn J, Vazirabad IY, Kong AL, Kriegel AJ, Liu Z, Kivelä RM, Murtagh G, Gutterman DD, Beyer AM.) JCI Insight. 2025 Nov 24;10(22) PMID: 41026534 PMCID: PMC12643535 SCOPUS ID: 2-s2.0-105022722049 09/30/2025
-
(Bikomeye JC, McGinley EL, Zhou Y, Tarima S, Kwarteng JL, Beyer AM, Yen TWF, Winn AN, Beyer KMM.) JACC Adv. 2025 Sep;4(9):102069 PMID: 40829368 PMCID: PMC12396096 SCOPUS ID: 2-s2.0-105013235004 08/20/2025
-
(Do Couto N, Hidde M, Grigoriadis G, Sparapani R, Durand M, Widlansky M, Jankowski C, Berendt M, Canales B, Golus S, Norwood Toro LE, Laud P, Kong A, Hoskins K, Lewandowski D, Phillips SA, Gutterman DD, Kriegel AJ, Beyer KMM, Beyer AM, Stolley M.) Cardiooncology. 2025 Aug 21;11(1):75 PMID: 40841694 PMCID: PMC12369258 SCOPUS ID: 2-s2.0-105014033548 08/22/2025
-
Autophagy modulates the mechanism of flow-mediated dilation upstream of telomerase
(Hughes WE, Hader SN, Astbury K, Brandt L, Ait-Aissa K, Gutterman DD, Beyer AM.) Basic Research in Cardiology. December 2025;120(6):1131-1140 SCOPUS ID: 2-s2.0-105021839661 12/01/2025
-
(Bikomeye JC, McGinley EL, Zhou Y, Tarima S, Kwarteng JL, Beyer AM, Yen TWF, Winn AN, Beyer KMM.) Cancer Surviv Res Care. 2025;3(1) PMID: 40881314 PMCID: PMC12382361 09/01/2025